Research
NF-kB
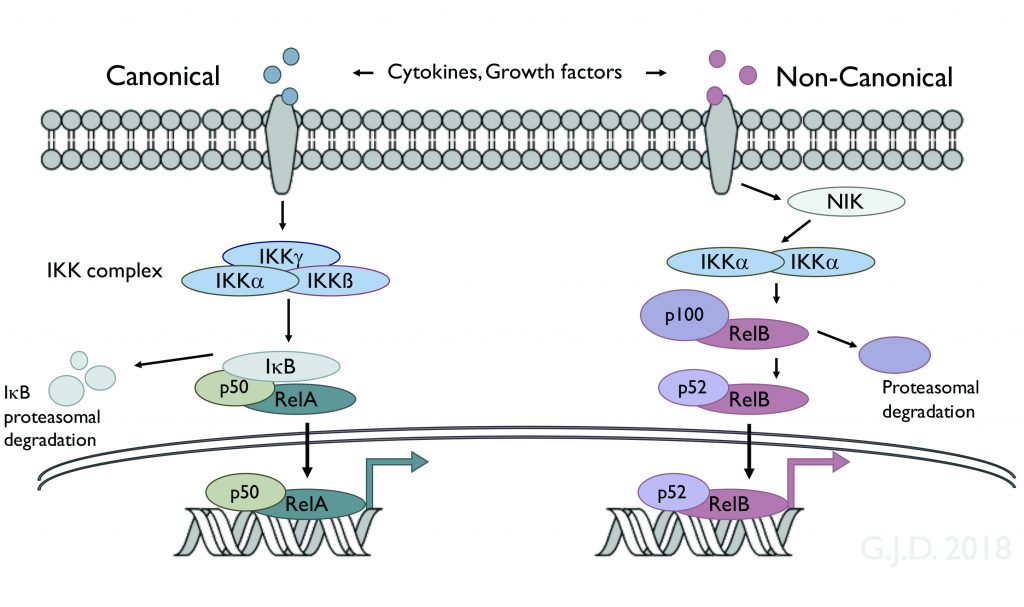
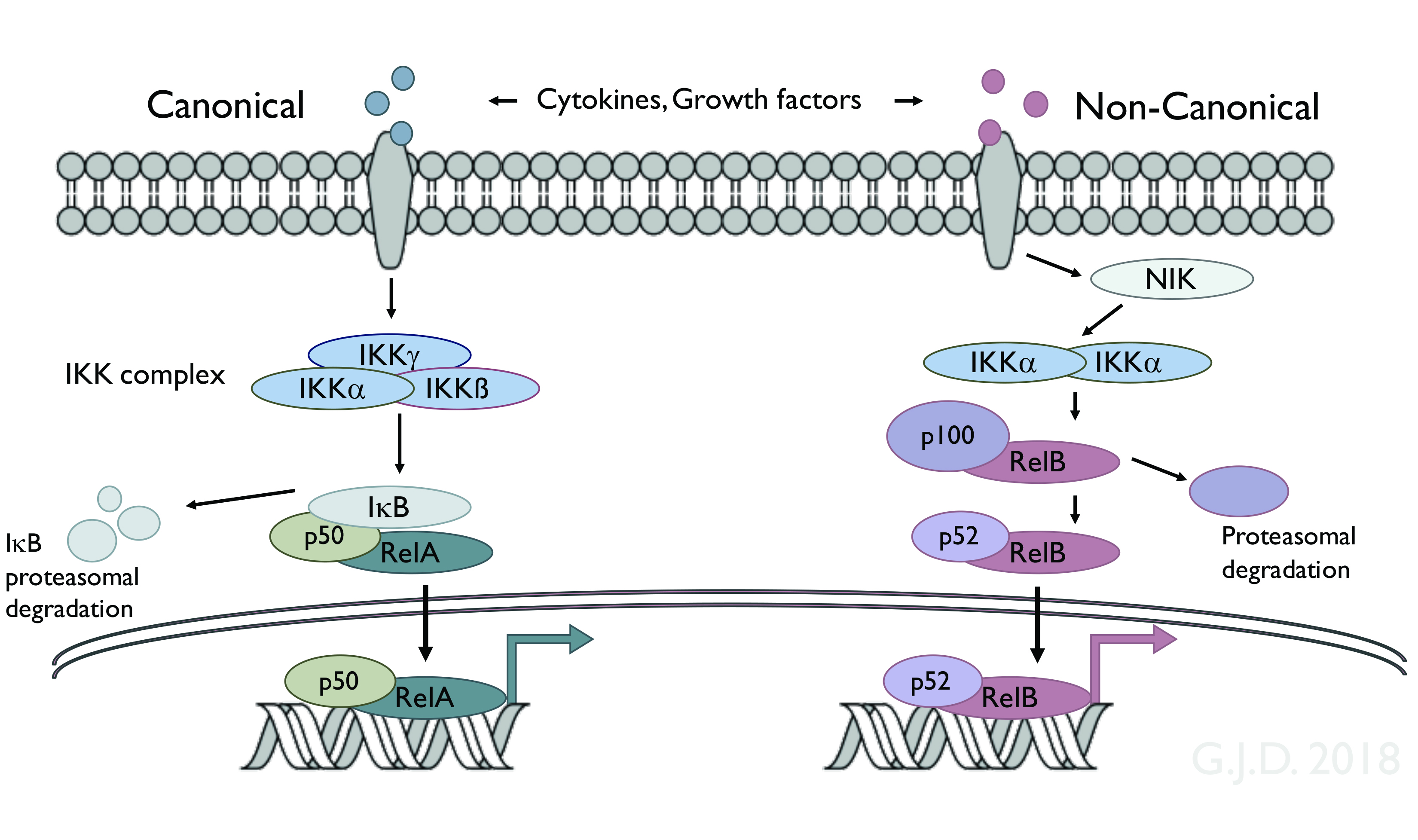
The laboratory continues to focus on the regulation and biological functions of the transcription factor NF-kappaB and on the role of NF-kappaB in disease, with a particular emphasis on cancer and cancer therapy. These studies focus on the signaling pathways which target NF-kappaB, on the biological mechanisms which NF-kappaB controls, and on the ability of NF-kappaB to promote basic oncogenic mechanisms as well as resistance to cancer therapy.
Recent Accomplishments
- NF-kappaB and control of cell growth and suppression of differentiation. We have shown that NF-kappaB promotes cell proliferation through the direct transcriptional regulation of the cyclin D1 promoter (published in Molecular and Cellular Biology). We have studied the link between cell growth and suppression of cell differentiation. Our data indicate that the activation of NF-kappaB in myoblasts suppresses the ability of these cells to differentiate into myofibers. The mechanisms for this suppression of differentiation comes from the ability of activated NF-kappaB to lead to the loss of the mRNA encoding MyoD. MyoD is the key transcription factor involved in muscle differentiation. Results in this paper indicated that the loss of MyoD mRNA induced by NF-kappaB occurred at a post-transcriptional level. This work was published in Science. Importantly, we have now found that another key transcription factor which controls differentiation of chondrocytes (Sox9) is lost by a similar mRNA-dependent mechanism (in press, Genes and Development). This study revealed that common motifs in the MyoD and Sox9 mRNAs control the NF-kappaB-dependent RNA loss. It is important to consider that the activation of NF-kappaB in certain cancers may inhibit tumor cell differentiation and, in this regard, we have begun the analysis of mRNAs that are downregulated in an NF-kappaB-dependent manner in certain cancers.
- Role of oncoprotein Bcl-3 in cell growth and oncoenesis. We have found that the IkappaB homologue Bcl-3 (cloned based on a translocation in certain leukemias) interacts with the NF-kappaB p52 subunit to strongly upregulate cyclin D1 gene transcription and to promote G1/S transition. This paper was published in Molecular and Cellular Biology. We believe that these findings are very important as previously we had described that Bcl-3 and p52 are upregulated in breast tumors (published in Oncogene) and it is known that cyclin D1 is also upregulated in breast cancer and may be required for tumor progression (at least in certain models). New experiments in the laboratory are focused on the potential involvement of Bcl-3 in controlling p53 function.
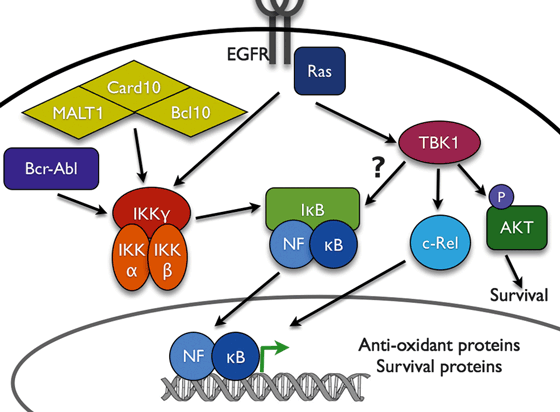
- Oncoprotein signaling and NF-kappaB: Ras- and BCR-ABL-induced pathways and NF-kappaB. We have previously shown that oncogenic Ras and oncogenic Raf activate NF-kappaB functional activity and that NF-kappaB is required for Ras to induce cellular transformation. Importantly we have shown that BCR-ABL requires Ras to induce NF-kappaB activation and that NF-kappaB is required for BCR-ABL to induce tumorigenesis (this paper was publised in Genes and Development and was the first to show the requirement of NF-kappaB in tumor formation). We have shown that Akt (downstream of the Ras effector PI3K) activates NF-kappaB through a mechanism independent of induced nuclear accumulation but dependent on the enhancement of inherent transcriptional potential (stimulating transactivation function). This process involves the IkappaB kinase, which is surprising since IKK was thought only to function to promote nuclear accumulation of NF-kappaB. This work was published at J. Biol. Chem. Another paper recently published in J. Biol. Chem. shows that Ras suppresses the traditional activation of NF-kappaB as induced by TNF (but not IL-1) while simultanenously promoting the activation of NF-kappaB-dependent gene expression. This work indicates that Ras induces NF-kappaB functional through a pathway which does not utilize the upstream signaling pathways associated with cytokine induction. The results also suggest the potential that Ras suppresses TNF induction of NF-kappaB in order to promote the chronic upregulation of JNK. In fact, we have published (Molecular and Cellular Biology) that NF-kappaB suppresses persistent JNK activation.
- NF-kappaB and suppression of apoptosis: new therapeutic approaches for cancer therapy. We have published in a series of papers (Science, Molecular and Cellular Biology) that NF-kappaB activation strongly blocks apoptosis through the upregulation of specific anti-apoptotic genes. We published in Nature Medicine that the inhibition of NF-kappaB through a gene therapy approach strongly sensitized tumors to chemotherapy. Furthermore, we have published in Cancer Research that systemic chemotherapy plus systemic NF-kappaB inhibition (with PS-341) strongly synergize to promote tumor cell apoptosis. Additionally, we have shown that thalidomide is a potent inhibitor of NF-kappaB (published at J. Biol. Chem.) and we have now data that thalidomide synergizes with chemotherapy in causes tumor regression through enhanced apoptosis.
- NF-kappaB and chromatin: novel findings regarding the involvement of IKK in controlling chromatin/histone modification. A paper from the laboratory was published in Molecular and Cellular Biology showing that NF-kappaB associates functionally with histone deacetylase proteins (HDAC1 and 2) and that NF-kappaB can actively promote transcriptional repression. Thus, NF-kappaB can both repress or activate gene expression according to promoter and inducer specificity. Recently, we published in Nature that the IKKa subunit of IKK associates with NF-kappaB-regulated promoters to control the phosphorylation of histone H3 on ser10. This finding is a key result in dissecting the factors which control modification of chromatin (according to the histone code hypothesis) to regulate gene expression. These studies are now being extended to determine a role of IKKa in controlling EGF-induced gene expression and the associated control of oncogenesis through EGF receptor family members.